
The first in a series of articles exploring each aspect of the pandemic in detail
By “Spartacus” – one of the authors of the orginal ‘Spartacus’ paper that went viral in 2021
The Severe Acute Respiratory Syndrome
There are five viruses in the genus Betacoronavirus that are known to infect humans: OC43, HKU1, SARS-CoV, MERS-CoV, and SARS-CoV-2. The alphacoronaviruses 229E and NL63 and the betacoronaviruses OC43 and HKU1 cause the common cold.
SARS-CoV, MERS-CoV, and SARS-CoV-2 are not the common cold. Their symptoms can range from cold or flu-like symptoms to an acute viral sepsis that leads to pneumonia, organ failure, and death.
Severe Acute Respiratory Syndrome Coronavirus (also known as SARS-CoV) was responsible for a major outbreak in Southeast Asia lasting from 2002 through 2004. The disease primarily affected people in China, Hong Kong, Taiwan, Singapore, and Vietnam, along with some healthcare workers in Canada. Relative to COVID-19, the outbreak was quite small. There were approximately 8100 to 8400 cases worldwide and nearly eight hundred fatalities.
In 2002, the first cases of SARS appeared in Guangdong province, China. Due to the severity of the virus’s pulmonary symptoms, it quickly drew attention from the authorities, who moved to treat and isolate patients as best they could. Many of these patients were put on ventilators given huge doses of methylprednisolone to keep them alive. Some survivors ended up with long-term health issues from this last-ditch treatment, including steroid-induced osteonecrosis.
In one particular incident, in the Amoy Gardens apartments in Kowloon, hundreds of people were infected with SARS when viral particles were transmitted between apartment units through the plumbing. The tenants there had allowed the traps in their bathroom floor drains to run dry, which caused aerosolized viral particles in human waste to rise up into their apartments, sickening them.
SARS was believed to have originated in bats, and was then transmitted to masked palm civets before being transmitted to humans as a zoonotic disease. However, its true origins remain unknown. Just as quickly as it appeared, it disappeared entirely, and no further cases were confirmed beyond 2004.
In 2012, the first case of Middle East Respiratory Syndrome was detected in Saudi Arabia. It was thought to have originated in bats, before hopping to camels, and then to people. MERS is a lethal disease, with a case fatality rate in the area of 30%, but it is thankfully quite rare, and this mortality figure may be an overestimate due to under-surveillance. MERS-CoV uses a different host entry receptor from SARS-CoV; Dipeptidyl Peptidase 4 (DPP4) instead of Angiotensin Converting Enzyme 2 (ACE2). This may contribute to some of its unique properties, vis a vis pathogenesis.
SARS-CoV is structurally and genetically very similar to SARS-CoV-2, the causative agent of COVID-19. Both viruses cause the same general set of symptoms. Dry cough, muscle aches, fever, and lethargy. In COVID-19, additionally, diarrhea, vomiting, and loss of sense of taste and smell have been reported. For the majority who contract COVID-19, these symptoms resolve in approximately one week, leaving one a little worse for wear, but otherwise, alive and healthy. In severe cases, both SARS and COVID-19 can proceed to a sepsis, pneumonia, atypical ARDS, organ failure, and death, due to a dysregulated and extreme over-exuberant immune response. COVID-19 additionally has some very unusual pathological properties, which will be discussed later.
The sequelae of SARS are very similar to those of COVID-19. In 2021, the media made numerous references to COVID-19 “long-haulers”, or people suffering from “long COVID”. Scientists refer to this condition as PASC, or Post-Acute Sequelae of COVID-19. This condition is not news to anyone who studied SARS; many SARS survivors from the 2002 to 2004 outbreak suffered long-term sequelae, including lung fibrosis and post-viral myalgic encephalomyelitis/chronic fatigue syndrome. Some of these people continued to experience ME/CFS for several years, as a matter of fact, with some survivors reporting being affected by SARS sequelae well into the 2010s.
The media acted like this so-called “Long COVID” was something mysterious. It is not. It is a well-established aspect of SARS-like viruses and has been recognized by the scientific literature for over a decade.
There were signs that SARS-CoV was a vascular illness, as well. Since it shares the same entry receptor with SARS-CoV-2, it follows that SARS-CoV may attack vascular endothelial tissue in much the same manner. Some papers dating as far back as 2005 describe some features of SARS-CoV as resembling a vasculitis.
How did COVID-19 blindside scientists so badly? What we at ICENI anecdotally observed was a sort of blank-slatism among researchers. Very few took the time to examine SARS pathology for clues, as we did. Instead, COVID-19 was treated as entirely novel. Many, many years of highly valuable SARS research were forsaken in favor of starting fresh.
It is arguable that the name COVID-19, itself, was misleading. Had the condition been called SARS-2, scientists and physicians may have been more inclined to examine SARS pathology to jump-start their research. Instead, they started from scratch. For a whole year, this left people scratching their heads. What was COVID-19, exactly?
The public, some panicking due to the media response, and some justifiably angry due to the lockdowns and other draconian control measures, had absolutely no idea what this virus even was, or how it made people so ill in the first place. Some began to suspect that they had been hoodwinked. Lots of people knew no one who’d died from it. Meanwhile, even a cursory examination of the primary journal sources shows thousands and thousands of papers being published on this disease and its observed properties on a continuous basis, which the media have consistently failed to report on in an accurate and forthright manner, leaving people with a deeply polarized impression of COVID-19 as somewhere between a harmless flu and smallpox. Some people are so flustered by dishonest coverage and waffling officials, they aren’t even sure if the virus actually exists or not.
The truth is somewhere in-between. It isn’t a flu, but it certainly isn’t smallpox, either.
Speaking of smallpox, for thousands of years, mankind coexisted with that incredibly deadly, nasty, disfiguring disease. Even though it sporadically caused outbreaks that killed millions of people, we did not purposely paralyze our economy, leaving people to deal with joblessness and deaths of despair, or take food out of the mouths of children, to counteract it. Although there were some reasonable epidemic control measures to deal with smallpox, for the most part, we simply lived our lives as normal and let nature take its course.
Now, naturally, the practice of modern medicine is inimical to this. That’s why there exist thousands of different kinds of drugs, vaccines, and other countermeasures to deal with various diseases. However, the frenzied, disorganized, and socioeconomically damaging overreaction to COVID-19 raises many questions of its own. Namely, what gives politicians, journalists, and billionaires the right to pretend to practice medicine?
Day in, day out, we are greeted by a procession of talking heads, like Bill Gates. Very few of them are scientists or doctors, and yet, they insist on us making massive lifestyle changes to suit their whims, while instructing average people to shun those who contradict the official narrative. Ethan Siegel, writing for Forbes, boldly stated that one must not “do their own research” when it comes to COVID-19.
This seems reasonable to a lot of people. After all, most people are regrettably not equipped with the knowledge necessary to grapple with epidemiology or virology on their own. However, it’s really beside the point. The real question here is, what would you actually see if you did happen to research COVID-19 on your own?
SARS-CoV-2
SARS-CoV-2 is an enveloped, positive-sense, ssRNA virus with a large genome around 29.8 kilobases long. Each individual SARS-CoV-2 virion is approximately spherical in shape and about 120 nanometers wide (for the sake of comparison, a typical strand of human hair is about 80,000 to 100,000 nanometers thick). Coronaviruses are named for their appearance under scanning electron microscopy, with a “halo” of Spike proteins that project from their surfaces.
Chemical & Engineering News – What do we know about the novel coronavirus’s 29 proteins?
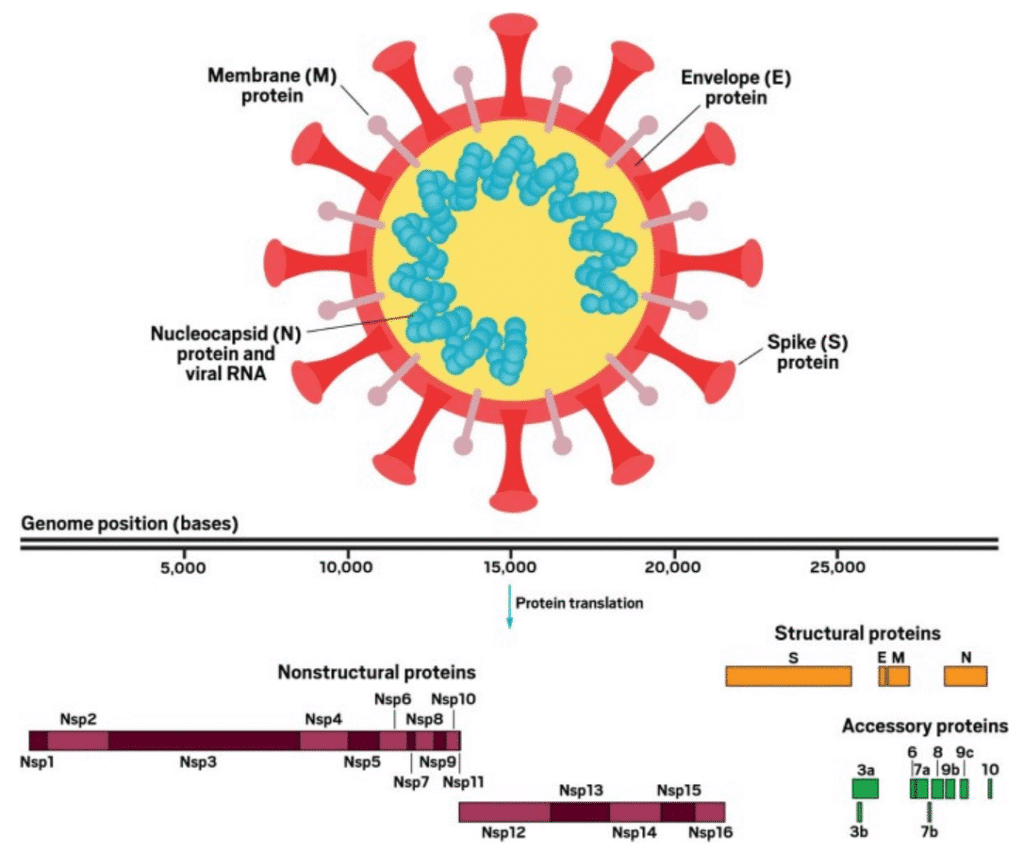
SARS-CoV-2 has four structural proteins (top): the E and M proteins, which form the viral envelope; the N protein (detail not shown), which binds to the virus’s RNA genome; and the S protein, which binds to human receptors. The viral genome consists of more than 29,000 bases and encodes 29 proteins (bottom). The nonstructural proteins get expressed as two long polypeptides, the longer of which gets chopped up by the virus’s main protease. This group of proteins includes the main protease (Nsp5) and RNA polymerase (Nsp12).
The virion has four structural proteins, S (Spike), E (Envelope), M (Membrane), and N (Nucleocapsid). The way the virus infects a cell is by latching its Spike protein to a receptor on the surface of a human cell, fusing the viral membrane and cellular membrane and/or drawing the virus into the cell by endocytosis. This process releases the coiled N protein containing the virus’s genome into the cell, which hijacks the cell’s protein synthesis machinery to make more virions.
Coronaviruses also produce double-membrane vesicles inside cells that behave as something akin to organelles, acting as factories for viral proteins.
Faculty Reviews – Membrane remodeling by SARS-CoV-2 – double-enveloped viral replication
The replicase complexes are found on double-membrane vesicles (DMVs) that contain viral double-stranded RNA. Expression of a small subset of viral proteins, including nsp3 and nsp4, is sufficient to induce formation of these DMVs in human cells, suggesting that both proteins deform host membranes into such structures. We will discuss the formation of DMVs and provide an overview of other membrane remodeling processes that are induced by coronaviruses.
Because some coronaviruses cause the common cold, some have been led to believe that COVID-19 is completely overblown; the common cold in disguise. However, not all coronaviruses are made equal, and the sarbecoviruses are universally nasty bugs.
COVID-19 Symptoms
COVID-19 is described in the medical literature as having “protean manifestations”, which is just a fancy way of saying that the symptoms vary widely. So widely, in fact, the disease frustrates diagnosis, mimicking a wide variety of other illnesses.
This is a list of many of the signs and symptoms attributed to COVID-19:
- Dry Cough
- Fever
- Headache
- Body aches
- Shortness of breath/Hypoxia
- Pneumonia and ARDS
- Coughing up flecks of blood
- Diarrhea
- Loss of sense of smell or taste, or altered smell or taste
- Meningoencephalitis
- Seizure (rarely)
- Dysautonomia
- Stroke
- Heart attack
- Pulmonary edema
- Pulmonary embolism
- Pulmonary fibrosis
- Heart muscle inflammation
- Kidney failure
- Intestinal hemorrhage
- Rash
- Purpura
- Transient livedo reticularis
- Diabetic ketoacidosis
- Testicular pain
- Sepsis
- Multiple organ failure
This is not even a comprehensive list.
Most express incredulity at all this. Can one single virus really cause such a wide variety of symptoms? Technically, yes. Particularly because it isn’t the virus itself doing most of it. It’s the patient’s own immune system being tricked by the virus into turning against their body. This will be described in further detail in later sections.
In the rare instances where it progresses to sepsis, COVID-19 can be a very serious, life-threatening illness. It has so many different manifestations, it can be difficult to diagnose, at first. That is, until one starts performing blood tests. COVID-19 is associated with a very peculiar set of lab results.
- O2 Sat <90%
- Elevated D-Dimer
- Elevated C-Reactive Protein
- Elevated IL-6 & TNF-alpha
- Normal Procalcitonin (elevated Procalcitonin may indicate severe disease)
- Elevated AST/ALT
- Elevated LDH
- Elevated Prothrombin Time
- Elevated Troponin
- Elevated Creatine Phosphokinase
- Thrombocytopenia
- Neutrophilia
- Lymphopenia
- Ferritinemia
- Hypokalemia
- Albuminuria
- Hematuria
If you see this on someone’s diagnostic test results, and they have a cough and hypoxia or pneumonia, you can almost be sure it’s COVID-19. A CT scan of the lungs and observation of ground-glass opacities will confirm it, however, some patients suffer from unusual “silent hypoxia” even without outward lung signs.
COVID-19 Clinical Course & Mortality
After exposure, it takes approximately 2 to 5 days for SARS-CoV-2 to incubate. After symptom onset, there are another 5 to 7 days of cold or flu-like symptoms. For the vast majority of infected, it resolves at this point, perhaps with some mild sequelae like persistent loss of smell or taste. For an unlucky few, it progresses to severe hyperinflammation at around day 8 to 10, and this lasts for around 10 or 11 days. The time from symptom onset to resolution or death can be as long as 21 days, approximately, with an additional 2 to 5 day incubation period preceding this.
COVID-19 can be divided into three distinct stages, beginning at the time of infection (Stage I), sometimes progressing to pulmonary involvement (Stage II, with or without hypoxemia), and less frequently to systemic inflammation (Stage III). In addition to modeling the stages of disease progression along with diagnostic testing, we have also created a treatment algorithm that considers age, comorbidities, clinical presentation, and disease progression to suggest drug classes or treatment modalities. This paper presents the first evidence-based recommendations for individualized treatment for COVID-19.
The reason why calculating SARS mortality during an ongoing pandemic is so difficult is because of the length of the disease’s course. It comes on in waves. As some people are dying, others are only just becoming infected, and so on. The only way to accurately tabulate mortality is to wait until the outbreak is over and then tally it up. Even so, there is a lot of data on COVID-19 mortality.
There are two different major figures used in epidemiology to examine the mortality of a disease. They are the Case Fatality Rate, or CFR, and the Infection Fatality Rate, or IFR. CFR is the number of confirmed cases of a disease divided by the number of deaths. IFR is the number of total infections from a disease divided by the number of deaths. The reason why they often prefer to use the CFR to measure the mortality of a disease is because many, many infections escape surveillance. The IFR is only ever a crude estimate, for the most part, even though it is, in essence, the true “mortality” figure of a disease.
For COVID-19, the IFR is approximately 0.23 to 0.27%, with a wide disparity in severity of illness dependent on age or comorbidities. This means that, on average, the virus kills approximately 1 in 434 to 1 in 370 people that it infects. In young and healthy people, the mortality is much, much lower than that. This is not Smallpox, and it’s not a death sentence. Far from it. It’s quite survivable for most.
Infection fatality rate of COVID-19 inferred from seroprevalence data
I included 61 studies (74 estimates) and eight preliminary national estimates. Seroprevalence estimates ranged from 0.02% to 53.40%. Infection fatality rates ranged from 0.00% to 1.63%, corrected values from 0.00% to 1.54%. Across 51 locations, the median COVID-19 infection fatality rate was 0.27% (corrected 0.23%): the rate was 0.09% in locations with COVID-19 population mortality rates less than the global average (< 118 deaths/million), 0.20% in locations with 118–500 COVID-19 deaths/million people and 0.57% in locations with > 500 COVID-19 deaths/million people. In people younger than 70 years, infection fatality rates ranged from 0.00% to 0.31% with crude and corrected medians of 0.05%.
Age is such a strong factor in COVID-19 morbidity and mortality, it has led to a vast gulf in the severity of the pandemic between wealthy nations with high average ages and impoverished ones with lots of young people and few elderly.
Nature – Age-specific mortality and immunity patterns of SARS-CoV-2
By contrast, for many European countries, we observe a higher incidence of deaths in older individuals than expected (Fig. 4a). This is consistent with the large proportion of reported COVID-19-associated deaths attributable to outbreaks in nursing homes, highlighting the enormous burden experienced by these communities in many higher-income countries22,23.
The severity of illness is highly variable, depending on a variety of comorbidities, particularly those involving endothelial dysfunction or pulmonary issues.
Overall, the severity of COVID-19 tends to be low on average.
Pathogenesis
SARS-CoV-2 uses a number of entry receptors to infect human cells, including Angiotensin Converting Enzyme 2 and Neuropilin-1, with GRP78, TMPRSS2, and Heparan Sulfate as contributing factors. However, the primary entry receptor is Angiotensin Converting Enzyme 2.
Angiotensin Converting Enzyme 2, or ACE2, is a part of the Renin-Angiotensin-Aldosterone System, or RAAS, which is a hormone feedback control system that moderates blood volume, vascular tone, and a variety of vascular inflammatory and tissue repair responses.
The way this system in the body works, Angiotensinogen from the liver is cleaved by Renin from the kidneys, producing the inactive Angiotensin I, which is further processed by Angiotensin Converting Enzyme to make the AT2 receptor agonist known as Angiotensin II. Angiotensin Converting Enzyme 2, in turn, acts to put the brakes on Angiotensin II by cleaving it to Angiotensin 1-7, which is a MAS receptor agonist. AT2 receptors and MAS receptors have opposite effects. AT2 receptors are vasoconstrictive and promote inflammation and oxidative stress, while MAS receptors are vasodilatory and anti-inflammatory and promote cell proliferation. Essentially, the body expresses more ACE2 if someone has high blood pressure, because it makes a vasoconstrictive hormone into its opposite, a vasodilator.
Because of the close relationship between the RAAS and the circulatory system, vascular endothelial cells and pericytes express a great deal of ACE2 enzymes. For those not familiar with biology, this requires a little bit of explanation. Genes are blueprints for proteins. Some cells express certain genes more than others, as a necessary aspect of their correct function. Many of the proteins produced by these genes become embedded in the cell’s plasma membrane, and are known as membrane-bound proteins. They perform vital functions, such as relaying signals from the exterior of a cell to its interior, or allowing certain molecules to pass through the membrane selectively. Angiotensin Receptors and Angiotensin Converting Enzymes found on the surfaces of vascular ECs are two of such membrane-bound proteins.
ACE2 is actually expressed in many different cell types in the body, including vascular endothelial cells and pericytes, airway and intestinal epithelial cells, brain astrocytes, renal tubules and podocytes, the seminiferous ducts of the testis, and more, which may explain SARS-CoV-2’s wide tropism in all of these tissues.
The way SARS-CoV-2 infects a cell is as follows:
SARS-CoV-2 Spike undergoes a conformational change whereby the trimeric heads of the Spike extend, latching onto ACE2 and locking firmly in place. Cell-surface Transmembrane Protease, Serine 2 (TMPRSS2) comes along and cleaves these heads off, exposing the “stalk” subunit of the protein underneath, which unfolds like an extension ladder, buries itself in the cell membrane, and then folds back on itself to draw the cell and the virus together, fusing them.
SARS-COV-2 uses a variety of cell lines as entry ports, particularly the epithelial cells of the airway.
Immune Network – SARS-CoV-2 Infection of Airway Epithelial Cells
The primary role of ACE2 is the maturation of angiotensin of the renin-angiotensin system, which controls blood pressure and vasoconstriction (23). ACE2 is expressed in the heart, blood vessels, kidney, esophagus, ileum, colon, upper and lower airways, cornea, liver, gallbladder, and testis (12). However, compared to other organs, the amount of gene or protein expression of ACE2 in the airways is low (12, 24). Still, entry of SARS-CoV-2 depends on the expression of receptors (ACE2, TMPRSS2, or cathepsin B and cathepsin L [CatB/L]) of the airways as the first gateway for the respiratory virus to initiate infection, and the distribution of receptors in the upper airway increases the infectivity of the virus (12). Originally, ACE2 plays a protective role in the acute lung injury in respiratory viral infections, such as SARS-CoV and influenza virus (25, 26, 27).
Olfactory epithelial sustentacular cells are another point of entry, and infection and injury of these cells may contribute to the anosmia experienced by COVID-19 sufferers.
Anosmia, the loss of smell, is a common and often the sole symptom of COVID-19. The onset of the sequence of pathobiological events leading to olfactory dysfunction remains obscure. Here, we have developed a postmortem bedside surgical procedure to harvest endoscopically samples of respiratory and olfactory mucosae and whole olfactory bulbs. Our cohort of 85 cases included COVID-19 patients who died a few days after infection with SARS-CoV-2, enabling us to catch the virus while it was still replicating. We found that sustentacular cells are the major target cell type in the olfactory mucosa. We failed to find evidence for infection of olfactory sensory neurons, and the parenchyma of the olfactory bulb is spared as well. Thus, SARS-CoV-2 does not appear to be a neurotropic virus. We postulate that transient insufficient support from sustentacular cells triggers transient olfactory dysfunction in COVID-19. Olfactory sensory neurons would become affected without getting infected.
Gastric epithelial cells are another possible point of entry, but it remains unconfirmed whether or not COVID-19 is transmissible by the oral-fecal route, or if it infects the GI tract through the bloodstream.
Frontiers in Immunology – Pathogenesis and Mechanism of Gastrointestinal Infection With COVID-19
However, whether SARS-CoV-2 can be transmitted through the fecal–oral route is still controversial. Infectious virus was isolated from intestinal tissue but not fecal specimens (16). Jeong et al. failed to directly prove the presence of viable SARS-CoV-2 in stool samples by cell culture isolation (17). Detection of high copy numbers of viral RNA in the stool does not equate to shedding of infectious viruses or transmission of the disease (18). Respiratory transmission was not specifically blocked, making it difficult to attribute the transmission to the fecal–oral route. Is the fecal viral load sufficiently high for human transmission? How long can the excreted virus persist in the environment? Can fecally shed virus infect animals that may serve as a reservoir for spread? During transmission, can gut be the first site of infection or does the virus spread to the gut from the respiratory or other tissues (18)? These all require more specific experimental demonstrations.
Due to the presence of large amounts of ACE2 receptors in the vascular endothelium, once inside the body, the virus has a strong affinity for blood vessels. It tries to have them for breakfast, as a matter of fact, leading to viremia and sepsis.
European Heart Journal – COVID-19 is, in the end, an endothelial disease
The initial characterization of COVID-19 as a pneumonitis incorporates the notion of disordered endothelial function. While initial infection of type I and II pneumocytes and alveolar macrophages no doubt participates in the initiation of infection, disordered endothelial function certainly contributes to the ongoing ravages of SARS-CoV-2 in the lung as elsewhere. Impaired endothelial barrier function can contribute to protein accumulation in the alveolar space and fluid accumulation and impaired oxygenation of the blood. IL-1 stimulation reduces VE-cadherin, dubbed the guardian of integrity of the endothelium. This finding links a cytokine storm directly to capillary leak, and aggravation of the adult respiratory syndrome (ARDS) picture that advanced COVID-19 presents.33,47 The deranged balance in the prothrombotic/antithrombotic properties of the endothelium can certainly contribute to thrombosis in situ in the pulmonary vasculature, as occurs in COVID-19.48 Impaired gateway function of the endothelium for traversal of leucocytes into tissues clearly participates in pneumonitis.
Chronic endothelial dysfunction does a number of undesirable things, including negatively affecting the balance of vasoconstrictors and vasodilators, and promoting oxidative stress that can lead to arterial hardening and atherosclerosis. However, the sepsis of COVID-19 leads to acute endothelial dysfunction. If one has pre-existing chronic endothelial dysfunction (i.e. metabolic syndrome), this stacks on top of that.
ACE2 has another function; it inactivates des-Arg9-bradykinin (DABK), an analog of the vasoactive peptide bradykinin. Bradykinin and the kallikrein-kinin system are closely related to the RAAS, but their function is not very well understood.
By fusing with and down-regulating ACE2, SARS-CoV-2 infection is thought to increase serum des-Arg9-bradykinin levels, leading to a “bradykinin storm”.
The newer bradykinin storm theory stresses the importance of the decreased angiotensin-converting enzyme 2 (ACE2) availability within the epithelial cells of the lungs, leading to an inability to degrade bradykinin analog, des-Arg9-BK within normal margins. ACE2 and bradykinin are both known components of the renin–angiotensin aldosterone system and now are uniquely tied into the pathophysiology of SARS–CoV-2.
Bradykinin and its receptors are involved in the condition known as hereditary angioedema, which is treated with an extremely expensive drug ($3,800 a dose) called Firazyr, which is the brand name for a solution containing the selective Bradykinin B2 Receptor antagonist known as icatibant.
SARS-CoV-2 has many other nasty tricks up its sleeve. Its E and 3a proteins are believed to act as calcium ion channels, a feature it shares with other coronaviruses. Coronaviruses need elevated intracellular calcium levels in order to replicate properly. It is a crucial part of their life cycle.
Evolution, Medicine, and Public Health – Conflicts over calcium and the treatment of COVID-19
Several recent studies have provided evidence that use of calcium channel blockers (CCBs), especially amlodipine and nifedipine, can reduce mortality from coronavirus disease 2019 (COVID-19). Moreover, hypocalcemia (a reduced level of serum ionized calcium) has been shown to be strongly positively associated with COVID-19 severity. Both effectiveness of CCBs as antiviral therapy, and positive associations of hypocalcemia with mortality, have been demonstrated for many other viruses as well. We evaluate these findings in the contexts of virus–host evolutionary conflicts over calcium metabolism, and hypocalcemia as either pathology, viral manipulation or host defence against pathogens. Considerable evidence supports the hypothesis that hypocalcemia represents a host defence. Indeed, hypocalcemia may exert antiviral effects in a similar manner as do CCBs, through interference with calcium metabolism in virus-infected cells. Prospective clinical studies that address the efficacy of CCBs and hypocalcemia should provide novel insights into the pathogenicity and treatment of COVID-19 and other viruses.
By using these proteins as viroporins to introduce calcium into the cells, the virus can supercharge its replication, however, it does this at the cost of stressing the cell severely. High intracellular calcium levels have a close relationship to reactive oxygen species formation by those cells.
Redox Biology – Calcium and ROS: A mutual interplay
Calcium is an important second messenger involved in intra- and extracellular signaling cascades and plays an essential role in cell life and death decisions. The Ca2+ signaling network works in many different ways to regulate cellular processes that function over a wide dynamic range due to the action of buffers, pumps and exchangers on the plasma membrane as well as in internal stores. Calcium signaling pathways interact with other cellular signaling systems such as reactive oxygen species (ROS). Although initially considered to be potentially detrimental byproducts of aerobic metabolism, it is now clear that ROS generated in sub-toxic levels by different intracellular systems act as signaling molecules involved in various cellular processes including growth and cell death. Increasing evidence suggests a mutual interplay between calcium and ROS signaling systems which seems to have important implications for fine tuning cellular signaling networks. However, dysfunction in either of the systems might affect the other system thus potentiating harmful effects which might contribute to the pathogenesis of various disorders.
Bradykinin receptors also have the ability to promote oxidative stress by the same pathways. These G-protein coupled receptors stimulate phospholipase C activity, which promotes intracellular calcium pathway activity, enhancing both viral replication and oxidative stress.
They also stimulate mitogen-activated protein kinase (MAPK), which promotes inflammation by way of enhanced transcription factor activity, such as by nuclear factor kappa B (NF-kB).
Also, bradykinin receptors upregulate prostaglandin levels by increasing cyclooxygenase (COX) pathway activity. In the presence of extreme oxidative stress, arachidonic acid in the COX pathway is oxidized into isoprostanes, which are oxidatively-produced prostaglandin-like compounds that are highly inflammatory.
Endothelial dysfunction is a hallmark of a wide range of cardiovascular diseases and is often linked to oxidative stress and inflammation. Our earlier study reported the formation of a functional heterodimer between bradykinin receptor 2 (B2R) and dopamine receptor 2 (D2R) that may modulate cell responses, dependent on intracellular signaling. Here, for the first time, we showed a cooperative effect of these receptors on the modulation of processes involved in oxidative stress, inflammation, and apoptosis in endothelial cells. Sumanirole, a specific D2R agonist, was shown to diminish the excessive production of reactive oxygen species induced by bradykinin, a proinflammatory B2R-activating peptide. This effect was accompanied by modified activities of antioxidant enzymes and increased phosphorylation of endothelial nitric oxide synthase, leading to enhance NO production. In turn, endothelial cell co-stimulation with B2R and D2R agonists inhibited the release of interleukin-6 and endothelin-1 and modulated the expression of apoptosis markers, such as Bcl-2, Bcl-xL, Bax, and caspase 3/7 activity. All these observations argue that the D2R agonist counteracts the pro-oxidative, pro-inflammatory, and pro-apoptotic effects induced through B2R, finally markedly improving endothelial functions.
…
Furthermore, BK can increase the release of F2-isoprostane in patients, leading to a strong pro-oxidative response in the human vasculature [13].
The increased phosphorylation of nitric oxide synthase may seem beneficial, at first. After all, among its many other functions, nitric oxide serves to scavenge reactive oxygen species such as superoxide. However, this is only true when endothelial nitric oxide synthase is coupled to its cofactor, tetrahydrobiopterin (BH4).
IUBMB Life – Tetrahydrobiopterin in nitric oxide synthase
Nitric oxide synthase (NOS) is a critical enzyme for the production of the messenger molecule nitric oxide (NO) from L-arginine. NOS enzymes require tetrahydrobiopterin as a cofactor for NO synthesis. Besides being one of the few enzymes to use this cofactor, the role of tetrahydrobiopterin in NOS catalytic mechanism is different from other enzymes: during the catalytic cycle of NOS, tetrahydrobiopterin forms a radical species that is again reduced, thus effectively regenerating after each NO synthesis cycle. In this review, we summarize our current knowledge about the role of tetrahydrobiopterin in the structure, function, and catalytic mechanism of NOS enzymes.
As it turns out, nitric oxide is actually antiviral against SARS-like coronaviruses. It inhibits the palmitoylation of the Spike protein (in other words, it inhibits palmitic acid attachment), which is a necessary step preceding fusion with ACE2.
Medical Sciences – Nitric Oxide: The Missing Factor in COVID-19 Severity?
First, NO resulted in the reduction of the palmitoylation of nascently expressed spike (S) protein, which affects the fusion between the S protein and its cognate receptor, angiotensin converting enzyme 2 (ACE2). Secondly, NO treatment of the virus resulted in a reduction in viral RNA production in the early steps of viral replication, and this could possibly be due to an effect on the two cysteine proteases encoded in Orf1a of SARS-CoV-1 (Figure 6).
This immediately tells us why COVID-19 is more severe in people with diabetes, hypertension, and obesity, and in the elderly. All of those groups have one thing in common; chronic endothelial dysfunction and reduced nitric oxide synthesis (and/or increased consumption by way of its reaction with ROS) and redox equilibrium issues as a result.
Current therapies for the comorbidities of metabolic syndrome, targetting nitric oxide and reactive oxygen species signaling in endothelial dysfunction. Metabolic syndrome is characterized by an increase in visceral adiposity, blood pressure, glucose intolerance, and dyslipidemia. Individually, these co-morbidities induce endothelial dysfunction by increasing reactive oxygen species (ROS) and reducing nitric oxide (NO; pathways indicated in black). ROS is increased via increases in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and pro-inflammatory adipokines and reductions in superoxide dismutase (SOD). This reduces endothelial nitric oxide synthase (eNOS) production via two key mechanisms: reduced L-arginine conversion and soluble guanylate cyclase (sGC) activity. Uncoupling of eNOS occurs via two mechanisms [tetrahydrobiopterin (BH4) and 5′-AMP-activated protein kinase (AMPK) inactivation] to further reduce eNOS activity. Increased cyclooxygenase-2 (COX-2) activity drives the production of vasoconstrictor prostanoids (PGF2a, prostaglandin F2α; TXA2, thromboxane A2) and decreases prostacyclin (PGI2) production. ROS also drives the production of other endothelium-derived contracting factors (ET-1= endothelin-1, 5-HT= serotonin and PE= phenylephrine).
However, there’s a big catch, here. We’ve already established that there are multiple events increasing intracellular Ca2+ pathway activity and oxidative stress. Therefore, these cells are going to be producing significant quantities of superoxide via NADPH oxidase.
When superoxide and nitric oxide react, they form a damaging nitrogen radical called peroxynitrite. Peroxynitrite, in turn, destroys the BH4 cofactor that endothelial nitric oxide synthase needs to make nitric oxide. When this happens, eNOS becomes uncoupled and starts producing more superoxide, instead of nitric oxide. This leads to a feedback loop. Superoxide and nitric oxide react to form peroxynitrite, peroxynitrite destroys the tetrahydrobiopterin cofactor in eNOS, eNOS produces superoxide, superoxide reacts with nitric oxide. This vicious cycle leads to the depletion of endothelial nitric oxide and the proliferation of damaging radicals.
Endothelial oxidative stress with ensuing decreased NO bioavailability appears as a likely pathogenic factor of endothelial dysfunction in ICU COVID-19 patients. A correlation between NO bioavailability and oxygenation parameters is observed in hospitalized COVID-19 patients. These results highlight an urgent need for oriented research leading to a better understanding of the specific endothelial oxidative stress that occurs during SARS-CoV-2.
Ordinarily, this is no problem. The body has ways of dealing with ROS, such as using enzymes to break it down. However, SARS-CoV-2 disables those enzymes.
To identify host factors or pathways important in the control of SARS-CoV2 infection, publicly available transcriptome data sets including transcriptome analysis of lung biopsies from COVID-19 patients were analyzed using differential expression analysis14. Here, genes linked with inflammatory and antiviral pathways, including RIG-I receptor and Toll-like receptor signaling, were enriched in COVID-19 patient samples, whereas genes associated with the NRF2 dependent antioxidant response were suppressed in the same patients (Fig. 1a–c). That NRF2-induced genes are repressed during SARS-CoV2 infections was supported by reanalysis of another data-set building on transcriptome analysis of lung autopsies obtained from five individual COVID-19 patients (Desai et al.15) (Fig. 1d). Furthermore, that the NRF2-pathway is repressed during infection with SARS-CoV2 was supported by in vitro experiments where the expression of NRF2-inducible proteins Heme Oxygenase 1 (HO-1) and NAD(P)H quinone oxydoreducatse 1 (NqO1) was repressed in SARS-CoV2 infected Vero hTMPRSS2 cells while the expression of canonical antiviral transcription factors such as STAT1 and IRF3 were unaffected (Supplementary Fig. 1). These data indicate that SARS-CoV2 targets the NRF2 antioxidant pathway and thus suggests that the NRF2 pathway restricts SARS-CoV2 replication.
We already know of a mechanism by which Nrf2 activity can reduce SARS-CoV-2 replication. The Nrf2 pathway breaks down ROS, which would restore endothelial health, which would lead to an abundance of nitric oxide release, which would suppress the virus’s Spike directly. It is in the virus’s own evolutionary interest to promote oxidative stress and suppress antioxidant pathways.
SARS-CoV-2 also suppresses and evades interferons, and its N protein directly promotes NLRP3 inflammasome activity. It also upregulates gene pathways associated with autophagy and promotes mitochondrial and endoplasmic reticulum stress and subsequent calcium dumping. SARS-CoV-2 also promotes significant syncytia formation.
This is, in a word, chaos.
The Consequences
This leads to a rather peculiar sequence of events. Afflicted cells begin spewing cytokines and ROS. The buildup of damage-associated molecular patterns, also known as DAMPs, also causes adjacent cells to detect these stress signals through their pattern recognition receptors (PRRs), which are like tiny smoke alarms on their surfaces that look specifically for these danger signals. This causes those cells to activate their transcription factors and start spewing cytokines, and so on and so forth.
Where there’s smoke, there’s fire. Patrolling leukocytes pick up these signals, and they get mad. Real mad.
The immune system is divided into two parts, generally speaking. The innate immune system, and the adaptive immune system. The purpose of the innate immune system is to suppress a pathogen long enough for the adaptive immune system to take over and neutralize it with antibodies.
“Suppress” is kind of an understatement when it comes to neutrophils and macrophages. Neutrophils actually start suicide-bombing the area with destructive enzymes that make literal peroxide and bleach (as in, more ROS, tragically enough), trying to denature and destroy pathogens. Macrophages clean up the hideous mess after them.
Normally, cells defend themselves from lipid peroxidation and the destruction of their membranes (which would precipitate cell death by ferroptosis and parthanatos) by aggressive neutrophil activity using enzymes like glutathione peroxidase to break down hydrogen peroxide into water and to convert lipid hydroperoxides into their corresponding alcohols. However, since SARS-CoV-2 suppresses the Nrf2 pathway, GPX is disabled and lipid hydroperoxides start building up instead. The body forms autoantibodies against these oxidized lipids, which it recognizes as foreign objects, and inflammation spirals out of control. Hyperferritinemia and a proliferation of Fenton reagent in the form of hydrogen peroxide and free iron leads to hydroxyl radical formation that starts damaging the tissues severely.
The process is something like this:
- SARS-CoV-2 promotes extreme intracellular calcium release while also suppressing Nrf2.
- Cells become extremely stressed. Endoplasmic reticulum and mitochondrial stress predominate.
- Superoxide is produced in large amounts.
- Superoxide reacts with nitric oxide to make peroxynitrite.
- Peroxynitrite uncouples nitric oxide synthase.
- Nitric oxide synthase releases more superoxide.
- Superoxide dismutase makes hydrogen peroxide from superoxide.
- Myeloperoxidase makes hypochlorous acid from hydrogen peroxide and chloride ions.
- Glutathione peroxidase fails to detoxify this.
- Hypochlorous acid starts stripping iron.
- Free iron, hydrogen peroxide, and superoxide make hydroxyl radicals via Haber-Weiss and Fenton reactions.
- Hydroxyl radicals oxidize lipids, leading to ferroptosis, parthanatos, and auto-antibody formation against oxidized lipids.
- Immune system goes in a feedback loop, seeing the DAMPs from this process and attacking the region with more ROS.
- Sepsis and death.
Essentially, most of the damage is caused by over-active neutrophils.
Furthermore, infiltrating neutrophils, a hallmark of COVID-19, can release myeloperoxidase (MPO), which can activate several pathways that lead to elevated cytokines and production of ROS such as hypochlorous acid (HOCl), superoxide (O2•-), and hydrogen peroxide (H2O2) 22–24. Notably, HOCl can both compete with O2 at hemoglobin heme binding sites and also cause heme degradation and subsequent release of free iron (Fe2+). Free iron can then undergo the Fenton reaction to produce an array of ROS, including the highly reactive hydroxyl radical (•OH) 23–27. Another possible facet of the observed pathophysiology in critical cases of COVID-19 is a decline in nitric oxide (NO), a key mediator of vasodilation 28, 29.
This proceeds in a positive feedback loop until the subject is suffering from acute sepsis, which promotes endothelial dysfunction, damages the glycocalyx lining their blood vessels, causes their capillaries to begin leaking into their lungs, while also disrupting blood chemistry, promoting coagulopathy and crowding out O2 from red blood cells.
The COVID-19 sufferer is by now walking into the ER, blue in the face, their lungs filling with tiny clots due to sepsis, and their red blood cells incapable of carrying oxygen because of aggressive ROS release.
Just What is Oxidative Stress, Anyway?
Reactive oxygen species are atoms or molecules that are missing a valence electron. This makes them “unhappy”. They want to replace that electron as quickly as possible, even if they have to steal it from things in their environment. This process of electron exchange is known as an oxidation-reduction reaction, or simply redox, for short.
Mitochondria are some of the main sites of redox activity in the body, but they’re not the only sites. There are many others.
Oxidation and reduction reactions happen all the time in living organisms. The Electron Transport Chain in mitochondria that makes ATP is absolutely vital to life, and yet, it involves oxygen, which is technically a rather reactive and damaging element on its own. After all, if you leave iron out on a table, maybe with a little water on it, you’ll get iron oxide. That is, rust.
People can rust, too. In the body, reactive oxygen species can react with lipids, DNA, and other stuff that you’re made out of, producing the oxidatively-modified counterparts of those molecules. The body does not like this, at all. Many different oxidized molecules in the body are treated as foreign objects by the immune system. They’re non-self. Debris and garbage.
You’ve undoubtedly heard about antioxidants in dietary supplements or health foods, without any further elaboration as to what antioxidants actually do. Antioxidants are molecules that sacrificially donate their electrons to radicals, so those radicals don’t have to steal them from more important molecules, like your cell walls or their genetic material.
Back in the day, when the famous chemist Linus Pauling found that oxidative stress was continuously damaging our lipids and DNA, he became a stalwart advocate of Vitamin C supplementation, which he believed would counteract this damage. However, oxidative stress in the body is not merely a destructive force. Many forms of reactive oxygen and nitrogen species are actually used by the body’s molecular signaling pathways, to trigger various kinds of activity in selective and surprising ways. And, in any case, as long as someone has plenty of antioxidant substrates in their body, like the kind one would get from a balanced diet, their cells’ own protective enzymes will act to moderate ROS levels to help maintain homeostasis.
Multi-Organ Dysfunction
COVID-19 does not only injure blood vessels in the lungs. It injures blood vessels and vital organs everywhere in the body.
COVID-19 can manifest as an intestinal disease, infecting the GI tract (this is where the notion of “anal swabbing” to test for COVID came from):
Although, as reported, SARS-CoV-2 infection mainly affects the respiratory system[32], leading to breathing difficulties, dry cough, and nasal congestion to respiratory failure, this novel coronavirus can be found in the GI tract as well[33]. Furthermore, SARS-CoV-2 RNA has been isolated from stools. Viral nucleocapsid protein can be observed in duodenal and rectal glandular epithelial cells by laser scanning confocal microscopy. The available results provide evidence of the activity of this virus in the GI tract[34].
COVID-19 can precipitate kidney failure:
Acute kidney injury (AKI) is common in patients hospitalized with coronavirus disease 2019 (COVID-19), reported in 24% to 57% of COVID-19 hospitalizations and 61% to 78% of intensive care unit admissions in patients with COVID-19.1–7 Compared with patients without COVID-19, those with COVID-19 develop more severe AKI, have greater dialysis requirements, and experience less in-hospital kidney recovery,2 which may increase their risk for incident chronic kidney disease (CKD) or progression of existing CKD.8
COVID-19 can lead to new-onset diabetes by infecting pancreatic islets and fat cells:
NEJM – New-Onset Diabetes in Covid-19
There is a bidirectional relationship between Covid-19 and diabetes. On the one hand, diabetes is associated with an increased risk of severe Covid-19. On the other hand, new-onset diabetes and severe metabolic complications of preexisting diabetes, including diabetic ketoacidosis and hyperosmolarity for which exceptionally high doses of insulin are warranted, have been observed in patients with Covid-19.1-3 These manifestations of diabetes pose challenges in clinical management and suggest a complex pathophysiology of Covid-19–related diabetes.
Nature – SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas
Infection-related diabetes can arise as a result of virus-associated β-cell destruction. Clinical data suggest that the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), causing the coronavirus disease 2019 (COVID-19), impairs glucose homoeostasis, but experimental evidence that SARS-CoV-2 can infect pancreatic tissue has been lacking. In the present study, we show that SARS-CoV-2 infects cells of the human exocrine and endocrine pancreas ex vivo and in vivo. We demonstrate that human β-cells express viral entry proteins, and SARS-CoV-2 infects and replicates in cultured human islets. Infection is associated with morphological, transcriptional and functional changes, including reduced numbers of insulin-secretory granules in β-cells and impaired glucose-stimulated insulin secretion. In COVID-19 full-body postmortem examinations, we detected SARS-CoV-2 nucleocapsid protein in pancreatic exocrine cells, and in cells that stain positive for the β-cell marker NKX6.1 and are in close proximity to the islets of Langerhans in all four patients investigated. Our data identify the human pancreas as a target of SARS-CoV-2 infection and suggest that β-cell infection could contribute to the metabolic dysregulation observed in patients with COVID-19.
Individuals infected with SARS-CoV-2 who also display hyperglycemia suffer from longer hospital stays, higher risk of developing acute respiratory distress syndrome (ARDS), and increased mortality. Nevertheless, the pathophysiological mechanism of hyperglycemia in COVID-19 remains poorly characterized. Here, we show that hyperglycemia is similarly prevalent among patients with ARDS independent of COVID-19 status. Yet among patients with ARDS and COVID-19, insulin resistance is the prevalent cause of hyperglycemia, independent of glucocorticoid treatment, which is unlike patients with ARDS but without COVID-19, where pancreatic beta cell failure predominates. A screen of glucoregulatory hormones revealed lower levels of adiponectin in patients with COVID-19. Hamsters infected with SARS-CoV-2 demonstrated a strong antiviral gene expression program in the adipose tissue and diminished expression of adiponectin. Moreover, we show that SARS-CoV-2 can infect adipocytes. Together these data suggest that SARS-CoV-2 may trigger adipose tissue dysfunction to drive insulin resistance and adverse outcomes in acute COVID-19.
COVID-19 even has neurological manifestations:
Nature – Post-acute neurological consequences of COVID-19: an unequal burden
The prevalence of neurological problems associated with COVID-19 in the acute and subacute phases of illness is 35–85% (Table 1)3,4,5. People commonly report cognitive or memory disturbances, headache, loss of smell or taste, and myalgia. Acute neurological diagnoses include encephalopathy, delirium, cerebrovascular disease, seizures, neuropathy and myopathy. Less frequently reported problems include abnormal movements, psychomotor agitation, syncope and autonomic dysfunction. Para-infectious complications, such as acute demyelinating encephalomyelitis, acute necrotizing encephalopathy, acute inflammatory demyelinating polyneuropathy and autoantibody-suspected neurological manifestations, have been documented in small retrospective studies, but data regarding their prevalence remain inadequate.
A lot of people scoffed at “COVID Toe” when they first heard of it, but it’s really just endothelial injury to small capillaries in the extremities:
A cutaneous condition called chilblains or COVID toes, which is associated with microvascular injury, has been diagnosed in children and young adults. Biopsies of these lesions have revealed endothelialitis with endothelial swelling and subendothelial infiltration of lymphocytes, lymphocytic vasculitis, and microthrombosis.21
In short, this is definitely not just a pneumonia.
How COVID-19 Is(n’t) Being Treated
The standard of care for this disease is laughable.
As per Peter McCullough, the standard operating procedure for COVID-19 is to do no early outpatient treatment at all. If someone comes into the ER complaining of flu-like symptoms and worrying that they have COVID-19, they’re told to go home and get bed rest, and they aren’t prescribed anything.
If they come back with sepsis, blue in the face, they’re proned and intubated almost immediately and given a steroid drip of dexamethasone, and maybe blood thinners like heparin to deal with the coagulopathy. They’re also given antivirals (much too late; the virus is already gone, it’s all just sepsis) and other futile therapies that do nothing to deal with the severe buildup of DAMPs that are activating the patient’s immune system in a vicious cycle.
Intubation and introduction of oxygen to hypoxic tissue mimics the physiology of ischemia-reperfusion injury. That is, it accelerates lipid peroxidation and oxidative stress by feeding cells with O2, the precursor of all ROS. Hypoxanthine and succinate breakdown leads to more superoxide release, which, in turn, leads to more lipid peroxidation, more DAMP accumulation, more neutrophil recruitment, and so on and so forth, injuring the tissues severely. The ROS makes those tissues steroid-insensitive, so the steroids stop working.
COVID-19 causes dry cough and lung fibrosis because it messes with bradykinin levels and because it promotes excessive monocyte-derived alveolar macrophage formation. It causes pneumonia, ARDS, sepsis, organ failure, coagulopathy, and angioedema because it attacks the lining of blood vessels and promotes capillary leak, and many leukocytes are recruited to the site of infection. It causes diabetes because it injures the islets of the pancreas and it reprograms fat cells. It causes skin manifestations because it attacks small capillaries serving up oxygenated blood to various features of the skin. It promotes kidney failure because the cells in renal tubules and podocytes express ACE2 just like blood vessels. The strokes, heart attacks, and PE are because it causes aggressive clotting. Again, many of its unusual features can be traced directly to its assault on the blood vessels. Even the so-called “silent hypoxia” COVID-19 is reported to cause can be more accurately described as a circulatory or blood chemistry issue rather than anything pertaining to lung physiology.
Anecdotally, one nurse I discussed this with stated that a patient of his needed to have both her legs amputated from the knees down. COVID-19 had caused such aggressive clotting in her legs, she lost all blood flow.
Another anecdotal report I saw involved a teenage Hispanic male in New York who suffered and died horribly as doctors tried desperately to balance the coagulopathy with hemorrhage, raising and lowering the dosage of anticoagulants they were giving him. Ultimately, he bled out through his intestines.
Basically, what they’re doing to these patients would be worse than doing nothing were it not for the fact that they’re critically desaturated and suffering from acute viral sepsis.
How COVID-19 Could Be Treated
Lots and lots of different ways.
- The first thing is, unironically, preventive diet and exercise. Having a balanced, micronutrient-rich diet, going on jogs, and getting adequate sunlight exposure all work to reverse endothelial dysfunction and chronic oxidative stress, making one’s blood vessels healthier and younger. This raises nitric oxide levels and balances the immune system, reducing the likelihood that one would get sepsis if they did happen to contract COVID-19, while also suppressing the replication of the virus. It also has the added benefit of increasing longevity and reducing health issues in general.
- Dietary supplements. N-acetylcysteine and glycine help raise and maintain glutathione levels, and together with selenium, they provide glutathione peroxidase the substrates necessary to detoxify ROS and lipid hydroperoxides. Vitamin D helps pump excess calcium out of cells, reducing NADPH oxidase activity and starving SARS-CoV-2 of the intracellular calcium it needs to replicate. Curcumin is a Nrf2 activator and raises GPX activity. Quercetin and Resveratrol are both potent antioxidants. Apocynin, one of the main constituents of Kutki Powder, is a powerful antioxidant that is known to moderate neutrophil activity, reducing myeloperoxidase’s formation of hypochlorous acid and reducing inflammatory cytokine activity.
- Dietary nitrate and/or inhaled nitric oxide. This may help to inhibit the virus’s Spike protein.
- Prophylactic antivirals. Most antivirals, such as Kaletra, Remdesivir, Ivermectin, and HCQ, should largely be given as post-exposure prophylaxis, because the viral load of COVID-19 tapers off to practically nothing after someone has been symptomatic for several days. That’s not what’s being done. What they’re actually doing in these clinical trials is giving antivirals to people who have sepsis, but no virus left in their bodies, and then claiming that antivirals don’t do anything, which is basically scientifically fraudulent. Additionally, Remdesivir and Kaletra are actually quite toxic.
- TMPRSS2 inhibitors. Camostat mesylate may prevent cleavage and activation of the Spike by TMPRSS2.
- Colchicine and allopurinol. Repurposed gout medication. The inhibition of hypoxanthine breakdown to xanthine and uric acid formation reduces oxidative stress and lipid peroxidation.
- Calcium channel blockers, like nifedipine and amlodipine. SARS-CoV-2 needs calcium to replicate, and excess intracellular calcium causes oxidative stress.
- Iron chelators, like deferoxamine. Unregulated free iron promotes hydroxyl radical formation and is highly pro-inflammatory and pro-oxidant.
- Methylene Blue. This may counteract the effects of excessive bradykinin.
- Repurposed drugs with antioxidant effects. Budesonide, famotidine, diphenhydramine, and fluvoxamine are all actually antioxidants, in addition to their typical effects. Additionally, histamine blockers such as the aforementioned Pepcid and Benadryl may help with mast cell overactivation in COVID-19 and the associated inflammation.
- Direct delivery of antioxidants. As in, literally pumping Vitamin C, glycine, glutathione, selenium, and N-acetylcysteine into people in intravenous form, or even delivering them with a nebulizer.
This is not even a comprehensive list, but it’s a start. I encourage people to examine the linked references for these substances and their potential uses as COVID-19 therapies. There are already many papers speculating on their use, as well as some ongoing clinical trials.
If people with COVID-19 were to be started on a cocktail of these things right away, as a form of early outpatient treatment, I’m willing to bet that most of them wouldn’t ever need a ventilator.
The Reckoning
Now, at this point, you may be wondering what on earth you just read. It doesn’t sound anything like what the media has described to the public, does it? You look in the news, and it’s page after page of pneumonia this, pneumonia that.
The reality of COVID-19 is that it is a far, far more complex syndrome than most people give it credit for, which has stumped hundreds of the brightest and best scientists on the planet, month after month.
The media have undersold COVID-19 as a “pneumonia”, while simultaneously overselling it as a new black plague, leading to public confusion, anger, and frustration. Only a few science-oriented outlets have even bothered to go into the precise mechanics of this virus.
Imagine if you didn’t “do your own research”. You would just have to take for granted that COVID-19 is a spooky pneumonia that sometimes causes blood clots and puts people in the ICU.
At some point, you have to wonder if you’re being deliberately kept in the dark.
Maybe, just maybe, there are some very evil people out there who want to keep this crisis going forever. Maybe they don’t want this disease to actually be treated successfully.

Categories: Breaking News, Did You Know?, Latest News, The Expose Blog, World News
I get paid more than $90 to $100 per hour for working online. I heard about this job 3 months ago and after joining this I have earned easily $10k from this without having online working skills . Simply give it a shot on the accompanying site…
Here is I started.…………>> https://Www.NETCASH1.Com
The best way to avoid problems is undoubtedly to prevent infection in the first place, and thus avoid post infection treatment, prophylaxis is good but only works after invasion by the pathogen. (personally, I use IVM, freely available here in Brazil) In conjunction I am now using PVP-I in a 0.5% concentration applied with a nasal atomizer to invalidate any airborne pathogen …… I am 70 years old and, so far so good ! This is how I prepare it :- https://www.facebook.com/media/set/?vanity=christopher.sellars.96&set=a.10166088150570026
I get paid more than $205 to $405 per HOUR for working online. I heard about this job 3 months ago and after joining this I have earned easily $30k from this without w having online working skills . Simply give it a shot on the accompanying site…
Here’s what I do…………>>> https://Www.SmartPay1.com
I get paid more than $90 to $100 per hour for working online. I heard about this job 3 months ago and after joining this I have earned easily $10k from this without having online working skills . Simply give it a shot on the accompanying site…
Here is I started.……… https://www.Easywork2.com/
All this and yet no one has presented a physical virus, a real one, in the flesh so to speak. Ever.
[BECOME A MEMBER] I make over $200-$300 an hour for online work. I heard about this job 3 months ago and after joining I easily made $30k with no online jobs knowledge. Just try it out on the attached page.
…
More details……. https://Www.WorkStar24.com
That is quite true and the REAL death rates in the UK are similar to those in a bad flu year so what is the point of all the waffle above? Learn to live with it and get on with your life.
> what is the point of all the waffle above
To generate money for the writer. They’re probably “university educated”. It sure as hell won’t make one iota of difference.
The point is that existing treatments may actually be making things worse.
The Substack is free, and all of my writings are released under Creative Commons. I haven’t accepted one red cent for any of this.
https://dailyexpose.uk/2021/12/03/new-study-proves-covid-is-a-fraud/
WTF is a “substack”? We don’t know. Everyone already knows that “existing treatments” are “making things worse” because jabbed people are dropping dead all over the place. All you’re doing is reiterating the same facts over and over and over again, and just as radio and tv killed publc health by making everyone spend all their lives sat on sofas, you’re killing all hope of any public uprising by making people read this stuff again and again instead of interacting with each other and rising up against their overlords. How about you write some “incitement to revolution” for a change?
Plus the mention of Remdesivir as if it’s one of the antivirals that should be given! Remdesivir is almost guaranteed to cause death!
The bad thing is that doctors should know how to treat this. Here is the description of multiple things that help. Instead, they let people get to the point where there is no more helping and they let them die. What kind of doctors are these?
Doctors that are getting paid £22.50 for every jab and therefore live in million pound houses with sports cars parked out the front.
A very good summary of all of the total crap science associated with this Op.
Ivermectin has been distributed by the WHO to infected areas as a treatment for parasitic infections for over 30 years. Especially in certain African countries, Mexico and India, it has been confirmed to be safe to distribute directly to people. In addition, ivermectin has been reported to suppress the invasion of SARS-CoV-2 into cells and inhibit replication. You can get your ivm by visiting https://ivmpharmacy.com
Here is a thought. How about you remove the “Would love your thoughts” symbol so it doesn’t show up on every single page when I try to print this article?
This article mentions Remdesivir as one of the antivirals that should be given patients early. Remdesivir is one of the primary causes of hospital-caused deaths in COVID patients! This should definitely be revised!